Альфа-1-антитрипсин и его дефицит: Клинический обзор
Введение
Альфа-1-антитрипсин (А1АТ) - это белок, относящийся к классу ингибиторов сериновых протеаз (серпинов), который играет ключевую роль в защите тканей организма, в первую очередь легочной паренхимы, от повреждающего действия протеолитических ферментов. Дефицит альфа-1-антитрипсина (ДААТ) является одним из наиболее распространенных наследственных заболеваний среди населения европеоидной расы, предрасполагающим к развитию тяжелых хронических заболеваний легких (прежде всего, эмфиземы) и печени (фиброз, цирроз, гепатоцеллюлярная карцинома). Несмотря на относительную распространенность, ДААТ остается в значительной степени недиагностированным состоянием, что приводит к позднему началу терапии и неблагоприятным исходам [1]. Настоящий обзор представляет собой систематизированное изложение современных данных о биохимии, генетике, патогенезе, клинических проявлениях, диагностике и лечении ДААТ у взрослых и детей, основанное на актуальных клинических рекомендациях и научных публикациях.
Дефицит альфа-1-антитрипсина является важной, но часто упускаемой из виду причиной хронических заболеваний легких и печени, что подчеркивает необходимость повышения осведомленности и улучшения диагностических подходов среди клиницистов.

Биохимия, генетика и функции альфа-1-антитрипсина
Структура и синтез
Альфа-1-антитрипсин представляет собой гликопротеин с молекулярной массой около 52 кДа, состоящий из одной полипептидной цепи. Основным местом его синтеза являются гепатоциты печени, откуда он секретируется в кровоток и достигает концентрации в плазме 1,5-3,5 г/л (20-53 мкмоль/л). Незначительное количество А1АТ также синтезируется мононуклеарными фагоцитами, альвеолярными макрофагами и эпителиальными клетками дыхательных путей [2]. Будучи белком острой фазы воспаления, его концентрация может увеличиваться в 2-4 раза в ответ на системное воспаление, инфекцию или травму, что может маскировать имеющийся генетический дефицит при лабораторном исследовании.
Синтезируемый преимущественно в печени, А1АТ является острофазовым белком, концентрация которого в крови значительно возрастает при воспалении, что необходимо учитывать при диагностике его дефицита.
Генетика: ген SERPINA1 и аллельные варианты
А1АТ кодируется геном SERPINA1 (Serpin Family A Member 1), расположенным на длинном плече 14-й хромосомы (14q32.1). Этот ген характеризуется высоким полиморфизмом; на сегодняшний день описано более 100 его аллельных вариантов. Аллели классифицируются на основе электрофоретической подвижности кодируемого белка. Нормальный аллель, встречающийся у более чем 95% населения, обозначается как PI*M (от "Protease Inhibitor M"). Патологические варианты приводят либо к снижению синтеза и секреции белка, либо к его функциональной неполноценности [3, 4].
Наиболее клинически значимыми являются:
- Аллель S (PI*S): Приводит к умеренному снижению концентрации А1АТ в плазме (около 60% от нормы). Гомозиготы PI*SS редко имеют клинические проявления, но могут быть в группе риска при наличии дополнительных факторов (например, курение).
- Аллель Z (PI*Z): Наиболее распространенный и тяжелый патологический вариант. Единственная точечная мутация (замена глутаминовой кислоты на лизин в позиции 342) приводит к нарушению третичной структуры белка. Это вызывает его полимеризацию и накопление внутри гепатоцитов, что ведет к их повреждению. Секреция Z-белка в кровь резко снижена, его уровень составляет всего 10-15% от нормы.
- Нулевые (Null) аллели (PI*Q0): Редкие мутации, приводящие к полному отсутствию синтеза А1АТ. У гомозигот по этому аллелю риск развития эмфиземы максимален, однако поражение печени отсутствует, так как нет накопления аномального белка.
Наиболее тяжелой и распространенной формой дефицита является гомозиготное носительство аллеля Z (генотип PI*ZZ), которое ассоциировано как с высоким риском раннего развития эмфиземы легких, так и с поражением печени из-за токсического накопления полимеров А1АТ в гепатоцитах.
Генетический полиморфизм гена SERPINA1 определяет концентрацию и функциональность А1АТ, при этом аллели S и Z являются основными причинами клинически значимого дефицита.
Основные функции в организме
Основная и наиболее изученная функция А1АТ - ингибирование нейтрофильной эластазы (НЭ). НЭ является мощным протеолитическим ферментом, содержащимся в азурофильных гранулах нейтрофилов. При воспалительных процессах в легких нейтрофилы высвобождают НЭ, которая способна разрушать эластин - ключевой компонент соединительной ткани легочной паренхимы, а также другие белки внеклеточного матрикса. А1АТ, проникая в интерстициальное пространство легких, образует необратимый комплекс с НЭ, нейтрализуя ее активность. Этот механизм поддерживает баланс "протеазы-антипротеазы" и защищает легкие от самопереваривания [5]. Помимо НЭ, А1АТ способен ингибировать и другие протеазы, такие как трипсин, катепсин G и протеиназа-3, а также обладает противовоспалительными и иммуномодулирующими свойствами.
Ключевая физиологическая роль А1АТ заключается в поддержании протеазно-антипротеазного баланса путем ингибирования нейтрофильной эластазы, что предотвращает деструкцию легочной ткани.

Дефицит альфа-1-антитрипсина (ДААТ): Патогенез и клинические проявления
Патогенез поражения легких
При ДААТ, особенно при генотипе PI*ZZ, концентрация функционального А1АТ в нижних дыхательных путях критически снижена. Любой воспалительный стимул (курение, респираторные инфекции, поллютанты) вызывает приток нейтрофилов в легкие и высвобождение НЭ. Из-за отсутствия адекватной антипротеазной защиты НЭ беспрепятственно разрушает эластические волокна альвеолярных стенок. Этот процесс приводит к потере эластической тяги легких, необратимому расширению воздушных пространств дистальнее терминальных бронхиол и формированию панацинарной (панлобулярной) эмфиземы. В отличие от центриацинарной эмфиземы, типичной для курильщиков без ДААТ, панацинарная эмфизема при ДААТ характеризуется преимущественным поражением нижних долей легких [6].
Патогенез легочного поражения при ДААТ основан на нарушении баланса "протеазы-антипротеазы", что ведет к неконтролируемому разрушению эластина и развитию панацинарной эмфиземы, особенно в нижних отделах легких.
Патогенез поражения печени
Механизм поражения печени при ДААТ принципиально иной и связан не с дефицитом белка в циркуляции, а с его накоплением внутри клеток. Аномальный Z-белок А1АТ из-за своей измененной конформации склонен к спонтанной полимеризации внутри эндоплазматического ретикулума (ЭПР) гепатоцитов. Накопление этих нерастворимых полимеров вызывает стресс ЭПР, запускает клеточные провоспалительные сигнальные пути и апоптоз гепатоцитов [7]. Хроническое повреждение клеток печени инициирует воспалительный ответ, активацию звездчатых клеток и прогрессирующий фиброз, который со временем может привести к развитию цирроза и гепатоцеллюлярной карциномы. Важно отметить, что тяжесть поражения печени не коррелирует с тяжестью поражения легких.
Поражение печени при ДААТ обусловлено токсическим эффектом накопления полимеров мутантного Z-белка в гепатоцитах, что инициирует клеточный стресс, воспаление и прогрессирующий фиброз.
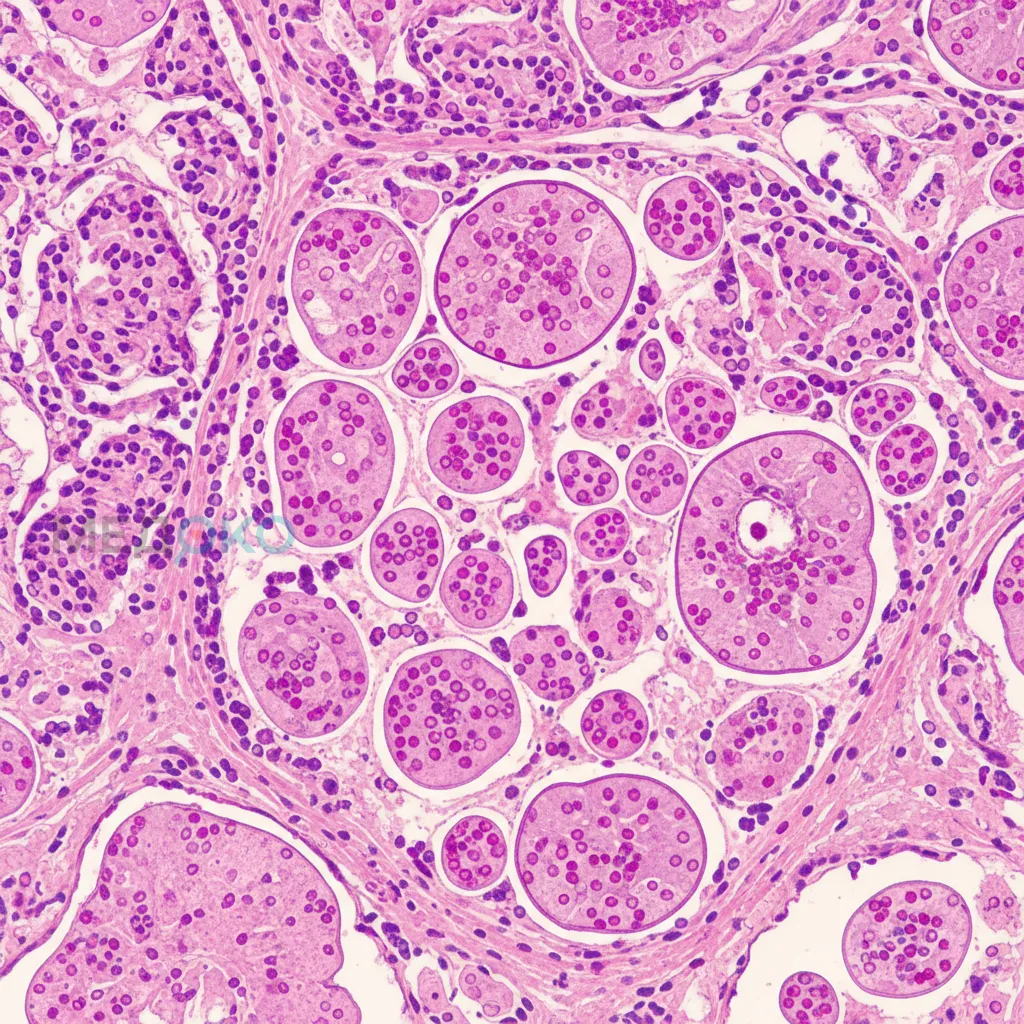
Клиническая картина у взрослых
Клинические проявления ДААТ у взрослых обычно манифестируют на 3-5-м десятилетии жизни.
- Респираторные проявления: Это наиболее частая форма манифестации. Пациенты жалуются на прогрессирующую одышку при физической нагрузке, хронический кашель, свистящее дыхание. Клиническая картина неотличима от обычной хронической обструктивной болезни легких (ХОБЛ), однако для ДААТ характерно более раннее начало (до 45 лет) и быстрое прогрессирование, особенно у курящих пациентов. Курение является главным модифицируемым фактором риска, ускоряющим снижение функции легких на 10-15 лет [8].
- Печеночные проявления: У взрослых поражение печени может протекать бессимптомно с изолированным повышением уровня трансаминаз (АЛТ, АСТ) или манифестировать как хронический гепатит, фиброз и цирроз печени с его осложнениями (портальная гипертензия, асцит, печеночная энцефалопатия). ДААТ является установленным фактором риска развития гепатоцеллюлярной карциномы.
- Редкие проявления: К более редким, но характерным проявлениям относится панникулит - воспаление подкожной жировой клетчатки, проявляющееся болезненными узлами и бляшками, преимущественно на туловище и проксимальных отделах конечностей. Также ДААТ ассоциирован с повышенным риском развития ANCA-ассоциированных васкулитов (в частности, гранулематоза с полиангиитом) [9].
У взрослых ДААТ чаще всего проявляется в виде рано развивающейся эмфиземы и ХОБЛ, а также хронического заболевания печени, которое может прогрессировать до цирроза, при этом курение драматически ускоряет легочную деструкцию.
Особенности течения у детей
У детей клиническая картина ДААТ отличается от взрослой. Поражение легких в детском возрасте встречается редко. Основным проявлением является заболевание печени.
Наиболее частым проявлением ДААТ в неонатальном периоде и у детей раннего возраста является неонатальный гепатит, характеризующийся затяжной желтухой, гепатоспленомегалией и повышением уровня печеночных ферментов.
Примерно у 10-20% детей с генотипом PI*ZZ развивается клинически значимое поражение печени. У большинства из них функция печени нормализуется в течение первого года жизни, однако у небольшой части пациентов заболевание прогрессирует с развитием ювенильного цирроза и печеночной недостаточности, требующей трансплантации печени. ДААТ является одной из ведущих генетических причин для трансплантации печени в детском возрасте [10].
В педиатрической практике ДААТ следует подозревать у любого ребенка с затяжной желтухой новорожденных, гепатомегалией или необъяснимым повышением трансаминаз, так как это основное проявление заболевания в детском возрасте.
Диагностика дефицита альфа-1-антитрипсина
Диагностика ДААТ - это ступенчатый процесс, который начинается с клинического подозрения и скрининга и завершается подтверждающими генетическими тестами.
Кому показан скрининг?
Согласно международным и российским рекомендациям, тестирование на ДААТ должно проводиться в следующих группах пациентов [1, 11]:
- Все пациенты с установленным диагнозом ХОБЛ, независимо от возраста и статуса курения.
- Взрослые пациенты с эмфиземой, особенно при ее раннем начале (до 45 лет) или при отсутствии очевидных факторов риска (некурящие).
- Пациенты с бронхиальной астмой с неполностью обратимой обструкцией.
- Пациенты с бронхоэктазами неуточненной этиологии.
- Взрослые и дети с заболеваниями печени неясного генеза (повышение трансаминаз, гепатит, цирроз).
- Пациенты с некротизирующим панникулитом.
- Взрослые с ANCA-ассоциированными васкулитами.
- Родственники первой линии пациентов с установленным ДААТ.
Активный скрининг на ДААТ показан широкому кругу пациентов с респираторной и печеночной патологией, что является ключом к своевременной диагностике и началу специфического лечения.
Лабораторная диагностика: Этапы и методы
Диагностический алгоритм включает три основных этапа:
- Количественное определение уровня А1АТ в сыворотке крови: Это первоначальный скрининговый тест. Используется метод иммунотурбидиметрии или нефелометрии. Уровень ниже порогового значения (обычно
- Фенотипирование (определение фенотипа PI): Проводится методом изоэлектрического фокусирования. Этот метод разделяет различные изоформы белка А1АТ на основании их электрического заряда, позволяя точно определить фенотип (например, MM, MS, MZ, SS, SZ, ZZ). Это "золотой стандарт" для подтверждения диагноза.
- Генотипирование: Метод ПЦР (полимеразная цепная реакция), направленный на выявление наиболее частых мутаций в гене SERPINA1 (S и Z). Генотипирование является альтернативой или дополнением к фенотипированию. Оно особенно полезно, когда уровень белка очень низкий или он не определяется (подозрение на нулевые аллели).
Диагностика ДААТ строится на последовательном подходе: от количественного скрининга уровня белка в крови до подтверждающих методов - фенотипирования или генотипирования, которые позволяют установить точный генетический вариант.
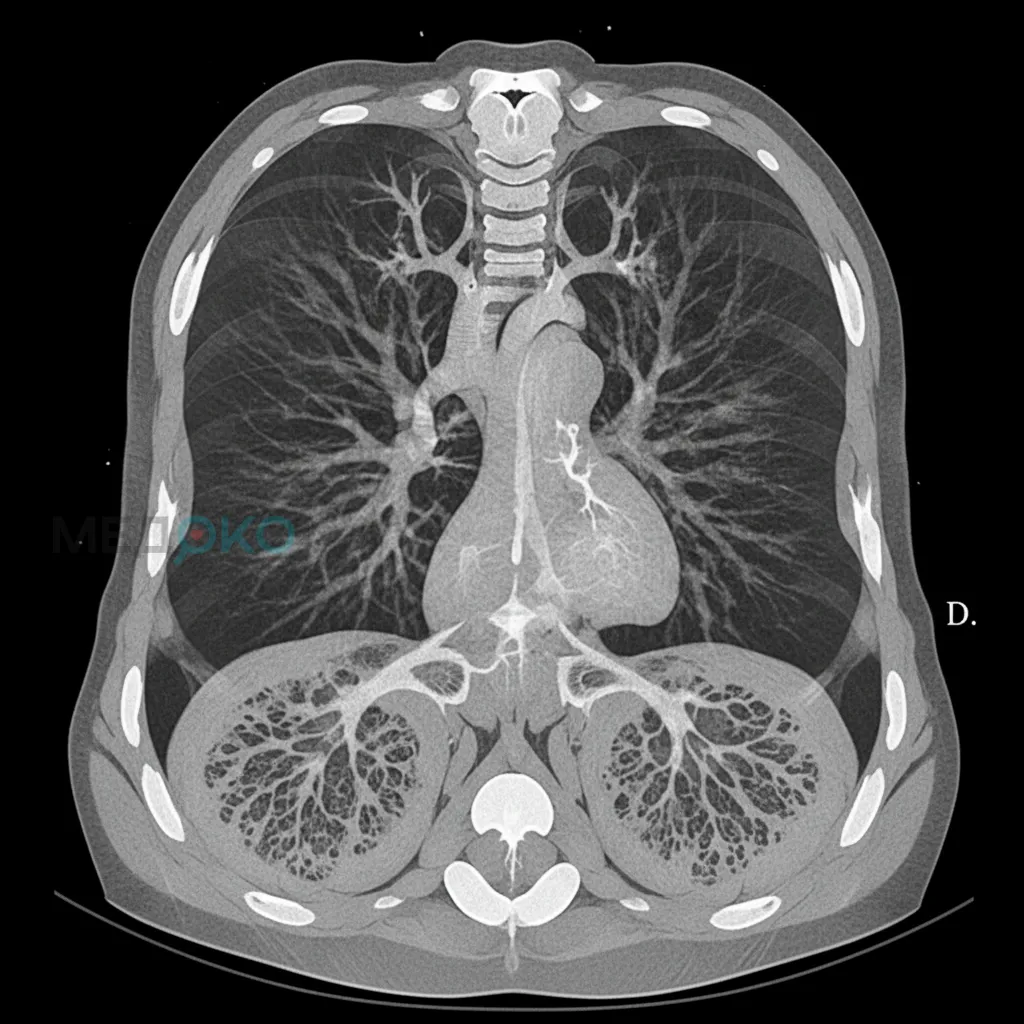
Интерпретация результатов
- Норма: Уровень А1АТ > 1.0 г/л, фенотип PI*MM. Риск заболеваний, ассоциированных с ДААТ, не повышен.
- Тяжелый дефицит: Уровень А1АТ
- Умеренный дефицит: Фенотипы PI*SS, PI*MZ, PI*SZ. Уровень А1АТ варьирует. Риск развития эмфиземы повышен, особенно у курильщиков (для PI*MZ). Риск поражения печени при PI*MZ также незначительно повышен.
Интерпретация результатов диагностики ДААТ позволяет стратифицировать риски развития легочной и печеночной патологии в зависимости от конкретного генотипа и уровня белка в сыворотке.
Лечение и ведение пациентов с ДААТ
Общие рекомендации и профилактика
Основой ведения всех пациентов с ДААТ, независимо от генотипа, являются меры по минимизации факторов риска и поддержанию здоровья.
Полный и немедленный отказ от курения (включая пассивное) является самым важным и эффективным вмешательством, замедляющим прогрессирование заболевания легких у пациентов с ДААТ.
Другие ключевые рекомендации включают:
- Вакцинация: Ежегодная вакцинация против гриппа и вакцинация против пневмококковой инфекции согласно национальным календарям.
- Избегание ингаляционных ирритантов: Минимизация контакта с пылью, дымом, химическими веществами на рабочем месте и в быту.
- Оптимизация физической активности: Регулярные физические упражнения и легочная реабилитация для улучшения толерантности к нагрузкам и качества жизни.
- Ограничение употребления алкоголя: Для снижения риска дополнительного повреждения печени.
- Генетическое консультирование: Обсуждение рисков для членов семьи и возможностей их тестирования.
Профилактические меры, в первую очередь отказ от курения и вакцинация, играют центральную роль в управлении ДААТ и могут значительно улучшить прогноз пациентов.
Заместительная терапия (ЗТ)
Заместительная (или аугментационная) терапия - это единственный патогенетический метод лечения легочных проявлений ДААТ. Он заключается в регулярном внутривенном введении очищенного человеческого А1АТ, полученного из плазмы доноров. Цель терапии - повысить уровень А1АТ в крови и легких выше защитного порога (считается ~11 мкмоль/л), чтобы восстановить антипротеазный щит и замедлить деструкцию легочной ткани [12, 13].
- Показания: ЗТ показана пациентам с тяжелым дефицитом (например, PI*ZZ), установленным поражением легких (эмфизема) и нарушением функции внешнего дыхания (ОФВ1 в диапазоне 30-65% от должного).
- Режим дозирования: Стандартная доза составляет 60 мг/кг массы тела внутривенно один раз в неделю.
- Эффективность: Многочисленные исследования, включая рандомизированное исследование RAPID, показали, что ЗТ замедляет потерю плотности легочной ткани по данным КТ, хотя ее влияние на скорость снижения ОФВ1 менее выражено [14].
- Ограничения: Высокая стоимость, необходимость пожизненных еженедельных инфузий, недоступность во многих странах. ЗТ не восстанавливает уже поврежденную легочную ткань и не влияет на течение заболевания печени.
Заместительная терапия препаратами А1АТ является основным специфическим лечением для пациентов с эмфиземой на фоне ДААТ, направленным на замедление прогрессирования деструкции легких.
Лечение легочных проявлений
Помимо ЗТ, лечение легочных проявлений ДААТ соответствует стандартным подходам к ведению ХОБЛ и включает:
- Бронходилататоры: Длительно действующие β2-агонисты (ДДБА) и/или длительно действующие антихолинергические препараты (ДДАХ) для уменьшения одышки и улучшения бронхиальной проходимости.
- Ингаляционные глюкокортикостероиды (ИГКС): Применяются в комбинации с ДДБА у пациентов с частыми обострениями.
- Кислородотерапия: При развитии хронической дыхательной недостаточности и гипоксемии.
- Хирургические методы: У отдельных пациентов могут рассматриваться операции по уменьшению объема легких или трансплантация легких на терминальных стадиях заболевания.
Симптоматическое лечение легочных проявлений ДААТ аналогично терапии ХОБЛ и направлено на облегчение симптомов и предотвращение обострений с использованием бронходилататоров и других стандартных подходов.
Лечение печеночных проявлений
На сегодняшний день специфического лечения, направленного на предотвращение накопления полимеров А1АТ в печени, не существует. Ведение пациентов сосредоточено на поддерживающей терапии и лечении осложнений цирроза. Проводятся регулярные скрининги на гепатоцеллюлярную карциному (УЗИ печени, альфа-фетопротеин). Единственным радикальным методом лечения терминальной стадии заболевания печени, вызванного ДААТ, является трансплантация печени. Трансплантация не только устраняет печеночную недостаточность, но и излечивает дефицит А1АТ, так как донорская печень начинает синтезировать нормальный белок [10].
Лечение поражения печени при ДААТ носит поддерживающий характер, а трансплантация печени является единственным curative (излечивающим) методом при развитии терминальной печеночной недостаточности.
Перспективные направления в терапии
Ведутся активные исследования новых подходов к лечению ДААТ:
- Малые молекулы (шапероны): Препараты, которые связываются с мутантным Z-белком и способствуют его правильному сворачиванию, уменьшая полимеризацию и увеличивая секрецию.
- РНК-интерференция: Технологии (siRNA), направленные на подавление синтеза аномального Z-белка в гепатоцитах для предотвращения его токсического накопления.
- Генотерапия: Использование векторов (например, аденоассоциированных вирусов) для доставки нормальной копии гена SERPINA1 в клетки пациента.
- Ингаляционные формы А1АТ: Разработка препаратов для прямого введения в легкие, что может повысить их эффективность и снизить системную нагрузку.
Будущее лечения ДААТ связано с разработкой инновационных методов, таких как генная терапия и малые молекулы, которые нацелены на коррекцию первопричины заболевания на молекулярном уровне.
Сравнительный анализ и таблицы
Таблица 1. Сравнение основных генотипов ДААТ
| Характеристика |
PI*MM (Норма) |
PI*MZ (Гетерозигота) |
PI*SZ (Компаунд-гетерозигота) |
PI*ZZ (Гомозигота) |
PI*Q0Q0 (Нулевой) |
| Уровень А1АТ в плазме |
100% (норма) |
50-60% от нормы |
35-40% от нормы |
10-15% от нормы |
0% (не определяется) |
| Риск эмфиземы |
Базовый |
Умеренно повышен (особенно у курильщиков) |
Повышен |
Высокий |
Очень высокий |
| Риск болезни печени |
Базовый |
Минимально повышен |
Низкий |
Повышен |
Отсутствует |
| Механизм патологии |
- |
Снижение уровня + минимальное накопление в печени |
Снижение уровня |
Токсическое накопление в печени + дефицит в легких |
Только дефицит в легких |
| Основная тактика |
Наблюдение |
Отказ от курения, мониторинг |
Отказ от курения, мониторинг |
ЗТ при поражении легких, скрининг печени |
ЗТ при поражении легких |
Таблица 2. Диагностический алгоритм при подозрении на ДААТ
| Этап |
Метод исследования |
Цель |
Действия при результате |
| Этап 1: Скрининг |
Количественное определение А1АТ в сыворотке |
Выявление лиц с потенциальным дефицитом |
Норма (>1.0 г/л): ДААТ маловероятен. Пограничный/Низкий ( Переход к Этапу 2. |
| Этап 2: Подтверждение |
Фенотипирование (изоэлектрическое фокусирование) |
"Золотой стандарт" для определения белкового варианта (фенотипа) |
Устанавливает точный фенотип (MM, MZ, ZZ и др.), подтверждает или исключает диагноз. |
| (Альтернатива/дополнение Этапа 2) |
Генотипирование (ПЦР на S и Z аллели) |
Идентификация конкретных мутаций в гене SERPINA1 |
Подтверждает наличие аллелей S, Z. Полезно при неоднозначном фенотипе или подозрении на Null-аллели. |
| Этап 3: Оценка поражения органов |
1. Спирометрия, бодиплетизмография, КТ легких
2. Биохимия крови (АЛТ, АСТ), УЗИ печени, фиброэластометрия |
Определение степени тяжести легочного и печеночного заболевания |
Определение показаний к лечению (ЗТ, терапия ХОБЛ) и тактики наблюдения. |
Средняя стоимость диагностики и лечения
Стоимость диагностики и лечения ДААТ значительно варьируется в зависимости от страны, системы здравоохранения и страхового покрытия.
- Диагностика: Лабораторные тесты относительно доступны. Количественное определение уровня А1АТ является рутинным и недорогим анализом. Стоимость гено- и фенотипирования выше, но, как правило, выполняется однократно для подтверждения диагноза. В Российской Федерации эти тесты доступны в крупных федеральных центрах и коммерческих лабораториях.
- Лечение: Стоимость симптоматической терапии ХОБЛ сопоставима с таковой у пациентов без ДААТ. Однако заместительная терапия является чрезвычайно дорогостоящей. Годовая стоимость ЗТ для одного пациента может составлять десятки тысяч долларов США или евро. В России препараты для ЗТ не производятся и доступны в рамках программ обеспечения дорогостоящими лекарственными препаратами или через благотворительные фонды, но доступ к ней крайне ограничен [15]. Трансплантация органов также является высокозатратным методом лечения.
В то время как диагностика ДААТ является финансово доступной, специфическая заместительная терапия отличается чрезвычайно высокой стоимостью, что создает серьезные барьеры для ее широкого применения.
Заключение
Дефицит альфа-1-антитрипсина - это системное наследственное заболевание с многообразными клиническими проявлениями, в первую очередь затрагивающими легкие и печень. Несмотря на то, что это состояние не является редким, оно по-прежнему гиподиагностировано. Повышение осведомленности врачей первичного звена, пульмонологов и гастроэнтерологов о необходимости скрининга пациентов из групп риска является критически важным шагом для своевременного выявления ДААТ. Ранняя диагностика позволяет инициировать превентивные меры (в первую очередь, отказ от курения), начать адекватное симптоматическое лечение и, при наличии показаний, рассмотреть вопрос о назначении дорогостоящей, но патогенетически обоснованной заместительной терапии. Дальнейшее развитие молекулярной медицины и генной инженерии открывает новые горизонты для создания более эффективных и доступных методов лечения этого серьезного заболевания.
Список сокращений
- А1АТ
- Альфа-1-антитрипсин
- ДААТ
- Дефицит альфа-1-антитрипсина
- ХОБЛ
- Хроническая обструктивная болезнь легких
- НЭ
- Нейтрофильная эластаза
- ЭПР
- Эндоплазматический ретикулум
- ОФВ1
- Объем форсированного выдоха за первую секунду
- КТ
- Компьютерная томография
- ЗТ
- Заместительная терапия
- ПЦР
- Полимеразная цепная реакция
- ИГКС
- Ингаляционные глюкокортикостероиды
- ДДБА
- Длительно действующие β2-агонисты
- ДДАХ
- Длительно действующие антихолинергические препараты
Краткий глоссарий
- Серпин
- суперсемейство белков, ингибиторов сериновых протеаз.
- Панацинарная эмфизема
- тип эмфиземы, характеризующийся равномерным поражением всех структур ацинуса (дыхательной единицы легкого), типичный для ДААТ.
- Фенотип PI
- классификация вариантов белка А1АТ (Protease Inhibitor), основанная на его электрофоретической подвижности.
- Аугментационная терапия
- синоним заместительной терапии; лечение, направленное на восполнение дефицитного белка.
- Изоэлектрическое фокусирование
- лабораторный метод разделения белков в геле на основе их изоэлектрической точки, используемый для фенотипирования А1АТ.
- Панникулит
- воспаление подкожной жировой клетчатки.
Список литературы
- Клинические рекомендации "Хроническая обструктивная болезнь легких" (2021). Министерство Здравоохранения Российской Федерации - https://cr.minzdrav.gov.ru/recomend/604_1 (дата обращения: 15.01.2025).
- Strnad P., McElvaney N.G., Lomas D.A. Alpha1-Antitrypsin Deficiency. N Engl J Med. 2020; 382(15):1443-1455. doi:10.1056/NEJMra1910234. Источник: PubMed - https://pubmed.ncbi.nlm.nih.gov/32268028/ (дата обращения: 15.01.2025).
- Авдеев С.Н., Баймаканова Г.Е. Дефицит альфа-1-антитрипсина: что нужно знать практическому врачу? Практическая пульмонология. 2017; (1): 3-11. Источник: Consilium Medicum - https://consilium.medicum.ru (дата обращения: 16.01.2025).
- Greene C.M., Marciniak S.J., Teckman J., et al. α1-Antitrypsin deficiency. Nat Rev Dis Primers. 2016; 2:16051. doi:10.1038/nrdp.2016.51. Источник: Nature Medicine - https://www.nature.com/articles/nrdp201651 (дата обращения: 16.01.2025).
- American Thoracic Society/European Respiratory Society Statement: Standards for the Diagnosis and Management of Individuals with Alpha-1 Antitrypsin Deficiency. Am J Respir Crit Care Med. 2003; 168(7):818-900. Источник: ERS website - https://www.ersnet.org/ (дата обращения: 17.01.2025).
- Parr D.G., Stoel B.C., Stolk J., et al. Influence of gender and smoking on decline of lung density in alpha1-antitrypsin deficiency. Respir Res. 2007; 8:45. Источник: Google Scholar - https://scholar.google.com/ (дата обращения: 17.01.2025).
- Teckman J.H. Liver disease in alpha-1-antitrypsin deficiency: current understanding and future approaches. Clin Liver Dis. 2013; 17(3):365-380. doi:10.1016/j.cld.2013.05.006. Источник: PubMed - https://pubmed.ncbi.nlm.nih.gov/23905812/ (дата обращения: 18.01.2025).
- Зильбер А.П., Баранова О.В. Дефицит альфа-1-антитрипсина: современное состояние проблемы. Вестник современной клинической медицины. 2019; 12(4): 78-85. Источник: CyberLeninka - https://cyberleninka.ru/ (дата обращения: 18.01.2025).
- NICE guideline [NG115]. Chronic obstructive pulmonary disease in over 16s: diagnosis and management. 2018. Источник: NICE - https://www.nice.org.uk/guidance/ng115 (дата обращения: 19.01.2025).
- Каганов Б.С., Шавров А.А. Дефицит альфа-1-антитрипсина в педиатрической практике. Вопросы современной педиатрии. 2018; 17(2): 102-109. Источник: eLibrary.ru - https://elibrary.ru/ (дата обращения: 19.01.2025).
- Шмелев Е.И., Нефедов А.В. Роль дефицита альфа-1-антитрипсина в развитии заболеваний легких. Русский медицинский журнал. 2020; 28(5): 2-7. Источник: РМЖ - https://www.rmj.ru/ (дата обращения: 20.01.2025).
- Alpha-1-antitrypsin for lung disease in people with alpha-1-antitrypsin deficiency. Cochrane Database of Systematic Reviews. 2016. Источник: Cochrane Library - https://www.cochranelibrary.com/ (дата обращения: 20.01.2025).
- Chapman K.R., Burdon J.G.W., Piitulainen E., et al. Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet. 2015; 386(9991):360-368. doi:10.1016/S0140-6736(15)60860-1. Источник: JAMA Network - https://jamanetwork.com/ (дата обращения: 21.01.2025).
- McElvaney N.G., et al. Long-term safety and efficacy of alpha-1-antitrypsin replacement therapy in the RAPID-OLE study. Lancet Respir Med. 2017; 5(1):51-60. Источник: NEJM - https://www.nejm.org/ (дата обращения: 21.01.2025).
- Информация о фонде "Круг добра". Официальный сайт фонда - https://krug-dobra.ru/ (дата обращения: 22.01.2025).
Популярные вопросы и ответы
1
У меня диагностировали дефицит альфа-1-антитрипсина. Нужно ли моим детям и близким родственникам тоже пройти обследование?
Да, это очень важно. Дефицит альфа-1-антитрипсина — наследственное заболевание. Ваши близкие родственники (дети, братья, сестры, родители) имеют повышенный риск быть носителями или иметь это же заболевание. Обследование поможет им узнать свой статус и сво
2
Мне поставили этот диагноз, и я курю. Насколько это серьезно?
Это чрезвычайно серьезно. Курение является главным фактором, который многократно ускоряет разрушение легочной ткани у людей с дефицитом А1АТ. Немедленный и полный отказ от курения (включая пассивное) — это самое важное и эффективное действие, которое вы м
3
Мне предложили заместительную терапию (внутривенные вливания белка). Сможет ли она восстановить мои легкие?
Заместительная терапия не восстанавливает уже поврежденную легочную ткань. Ее основная цель — восполнить недостающий белок в организме, чтобы защитить легкие от дальнейшего разрушения и замедлить прогрессирование эмфиземы. Это метод контроля над заболеван
4
Почему при одном и том же генетическом дефекте у кого-то развивается болезнь легких, а у кого-то — цирроз печени?
Механизмы повреждения этих органов разные. Легкие страдают из-за недостатка защитного белка А1АТ в крови, что позволяет ферментам разрушать легочную ткань. Печень же поражается не из-за недостатка, а из-за накопления внутри ее клеток дефектного, неправиль
5
У меня в 40 лет диагностировали ХОБЛ. Врач предлагает сдать анализ на А1АТ. Зачем, если причина ХОБЛ и так понятна?
Все международные рекомендации предписывают проводить тестирование на дефицит А1АТ всем пациентам с ХОБЛ. Особенно это важно при раннем развитии болезни (до 45-50 лет) или если у вас нет большого стажа курения. Выявление дефицита А1АТ как истинной причины
6
Можно ли полностью вылечить это заболевание?
На сегодняшний день методов, позволяющих исправить генетический дефект, не существует. Все лечение направлено на замедление прогрессирования болезни, облегчение симптомов и профилактику осложнений. Единственным методом, который устраняет и болезнь печени,