ВНИМАНИЕ: ЭКСТРЕННАЯ СИТУАЦИЯ!
Синдром Тюрко связан с высоким риском развития опухолей головного мозга и рака толстой кишки. Немедленно вызывайте скорую помощь (103 или 112), если у вас или вашего близкого внезапно развились: судорожный припадок (даже если он первый в жизни), потеря сознания, неукротимая утренняя рвота на фоне сильной головной боли (признаки отека мозга), примесь алой крови в кале в большом объеме или черный, дегтеобразный стул (признаки кишечного кровотечения).
За 30 секунд (Главное о болезни)
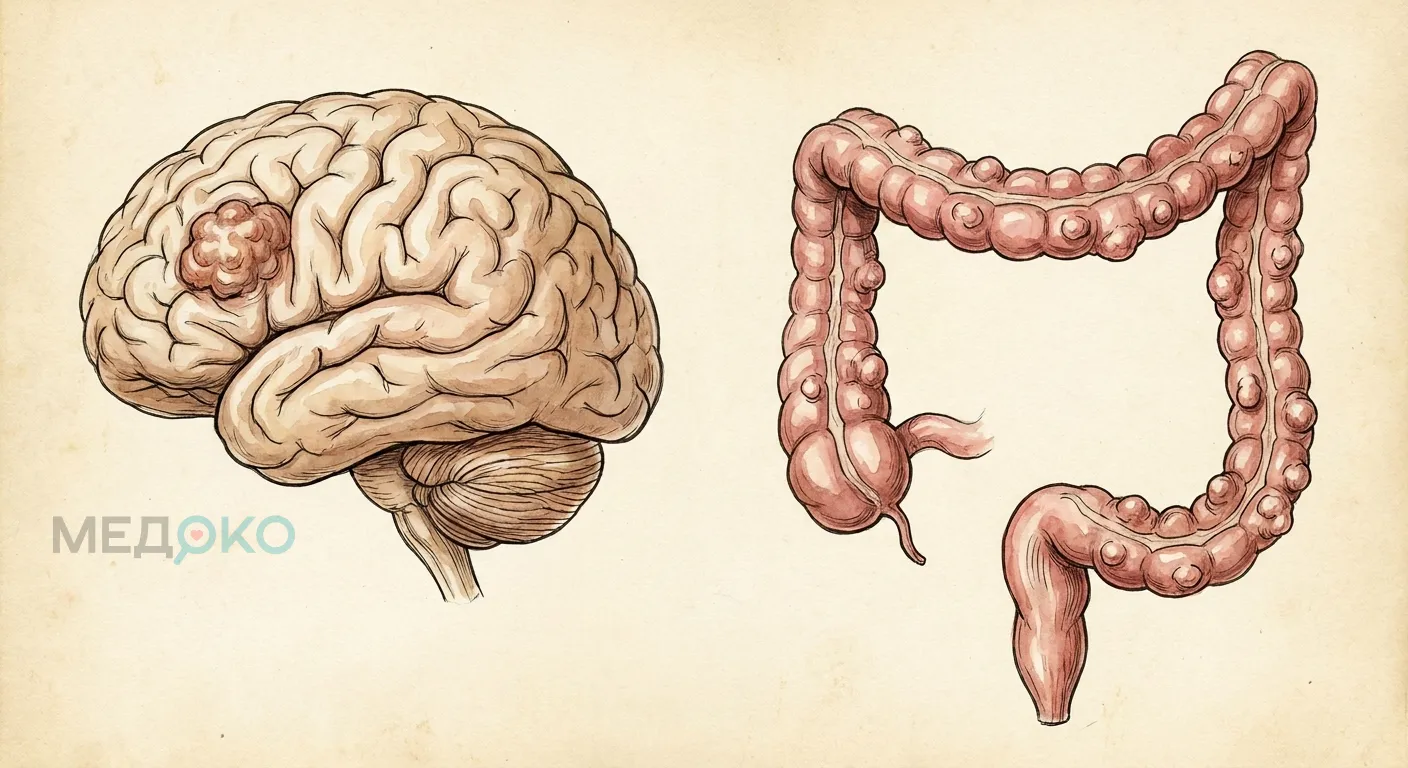
Что это: Редкое наследственное заболевание, при котором у пациента одновременно развиваются множественные полипы (или рак) толстой кишки и первичная опухоль головного мозга.
Причина: Генетические мутации, передающиеся по наследству (гены APC или гены системы репарации ДНК - MLH1, PMS2).
Код МКБ-10: Единого кода нет. Часто шифруется как Q85.8 (Другие факоматозы), D12.6 (Доброкачественное новообразование ободочной кишки), в сочетании с кодами опухолей мозга (С71).
Сколько длится: Заболевание обусловлено генетически и сохраняется пожизненно, требуя постоянного контроля.
Главное правило пациента: Регулярный тотальный скрининг - колоноскопия и МРТ головного мозга по строгому графику, не дожидаясь появления симптомов.
К какому врачу обращаться: Врач-генетик, гастроэнтеролог, онколог, нейрохирург, невролог.
Оглавление
РАЗДЕЛ 1 - ЧТО ТАКОЕ БОЛЕЗНЬ
Синдром Тюрко (Turcot syndrome) - это редкий генетический синдром, характеризующийся сочетанием первичных опухолей центральной нервной системы (чаще всего глиобластомы или медуллобластомы) и аденоматозного полипоза толстой кишки, который обладает крайне высоким риском злокачественного перерождения (колоректального рака) [1].
По сути, синдром Тюрко не является отдельным самостоятельным заболеванием в современном генетическом понимании. Врачи рассматривают его как специфический и тяжелый вариант течения двух других известных наследственных патологий: семейного аденоматозного полипоза (САП) и синдрома Линча (наследственного неполипозного колоректального рака).
Сравнительная таблица: Синдром Тюрко vs похожие диагнозы
| Признак | Синдром Тюрко | Семейный аденоматозный полипозом (САП) | Синдром Линча | Синдром Гарднера |
|---|---|---|---|---|
| Опухоли кишечника | Множественные полипы или ранний рак | Тысячи полипов кишки | Мало полипов, высокий риск рака | Тысячи полипов кишки |
| Внекишечные проявления | Опухоли головного мозга | Редко (в основном только кишка) | Рак матки, яичников, желудка | Остеомы (опухоли костей), опухоли мягких тканей |
| Возраст начала (в среднем) | Детство / Молодой возраст | Подростковый возраст | 30-50 лет | Подростковый / Молодой возраст |
Как отличить от синдрома Гарднера: Оба синдрома являются вариантами мутации гена APC. Ключевое отличие заключается в локализации внекишечных опухолей. Если у пациента с полипозом появляются костные наросты (остеомы, часто на челюсти) или фибромы кожи - это синдром Гарднера. Если неврологическая симптоматика и на МРТ выявляется опухоль мозга - это синдром Тюрко.

Ключевые выводы:
- 1. Синдром Тюрко - это смертельно опасная связка «полипоз/рак кишечника + опухоль мозга».
- 2. Синдром является не отдельной болезнью, а тяжелым фенотипическим проявлением синдрома Линча или САП.
- 3. Без превентивного хирургического лечения и скрининга риск летального исхода в молодом возрасте стремится к 100%.
РАЗДЕЛ 2 - ПРИЧИНЫ И ФАКТОРЫ РИСКА
Основная и единственная причина развития синдрома Тюрко - врожденные генетические мутации, которые пациент получает от родителей. Болезнь не вызывается плохим питанием, стрессом или инфекциями, хотя образ жизни может незначительно влиять на скорость малигнизации (озлокачествления) полипов.
Механизм развития связан с поломкой генов, которые в норме должны подавлять рост опухолей (гены-супрессоры) или исправлять ошибки в ДНК при делении клеток (гены системы мисматч-репарации). Когда эти гены не работают, клетки кишечника и клеток-предшественников в головном мозге начинают бесконтрольно делиться [2].
Таблица факторов риска (по группам):
| Группа факторов | Описание и влияние |
|---|---|
| Генетические (главный фактор) | Наличие мутаций в гене APC (тип 2) или генах MLH1, PMS2, MSH2, MSH6 (тип 1). Передается по наследству (чаще аутосомно-доминантно). |
| Семейный анамнез | Наличие у близких родственников рака кишечника до 50 лет или опухолей мозга. |
| Поведенческие / Образ жизни | Диета, богатая красным мясом и бедна клетчаткой, курение (ускоряют трансформацию уже имеющихся полипов в рак, но не вызывают сам синдром). |
Внимание: Если у одного из родителей подтвержден синдром Тюрко (или мутации САП/Линча), вероятность передачи дефектного гена ребенку составляет 50% при каждой беременности (при аутосомно-доминантном типе наследования).
Ключевые выводы:
- 1. Болезнь имеет исключительно генетическую природу.
- 2. Наличие родственников с ранним раком кишки или опухолями мозга - повод для немедленной консультации генетика.
- 3. Образ жизни не может предотвратить появление полипов при мутации, но может повлиять на скорость их роста.
РАЗДЕЛ 3 - КЛАССИФИКАЦИЯ И ФОРМЫ
В современной медицинской практике синдром Тюрко принято делить на два основных типа в зависимости от генетической мутации и вида опухоли мозга. Это разделение критически важно, так как оно определяет тактику лечения и прогноз [3].
Тип 1 (Ассоциированный с синдромом Линча)
- Генетика: Мутации в генах системы репарации ДНК (чаще всего MLH1 или PMS2).
- Опухоль мозга: Как правило, агрессивная глиома (глиобластома).
- Поражение кишечника: Полипов относительно немного (обычно менее 100, часто единичные), они крупнее и крайне быстро перерождаются в колоректальный рак.
- Сроки манифестации: Опухоли чаще выявляются во взрослом возрасте (в среднем в 30-50 лет), хотя бывают и более ранние случаи.
- Тактика: Регулярная колоноскопия (удаление полипов), при развитии рака - сегментарная или тотальная колэктомия. МРТ головного мозга.
Тип 2 (Ассоциированный с семейным аденоматозным полипозом - САП)
- Генетика: Мутация в гене APC.
- Опухоль мозга: Чаще всего медуллобластома (опухоль мозжечка).
- Поражение кишечника: Сотни и тысячи мелких полипов, устилающих всю толстую кишку (ковровый полипоз).
- Сроки манифестации: Развивается в детском и подростковом возрасте. Пик выявления медуллобластомы - до 20 лет. Рак кишки почти неизбежен к 30-40 годам.
- Тактика: Превентивное (профилактическое) полное удаление толстой кишки в молодом возрасте. Контроль состояния головного мозга.

Ключевые выводы:
- 1. Форма синдрома определяется не симптомами, а генетическим тестом.
- 2. Тип 1 связан со взрослыми глиобластомами и небольшим числом полипов.
- 3. Тип 2 поражает детей/подростков (медуллобластома) и вызывает тотальный полипоз кишки.
РАЗДЕЛ 4 - СИМПТОМЫ И ПРИЗНАКИ
Клиническая картина синдрома Тюрко складывается из двух независимых групп симптомов: неврологических и гастроэнтерологических. Они могут появиться одновременно, но чаще одна группа симптомов опережает другую на несколько лет.
Гастроэнтерологические симптомы (кишечные):
Сами по себе полипы долгое время бессимптомны. Признаки появляются при их изъязвлении, множественном росте или перерождении в рак:
- Хроническая диарея или, наоборот, стойкие запоры (смена характера стула).
- Примесь слизи и крови в кале (от скрытой крови до видимых сгустков).
- Схваткообразные боли в животе.
- Анемия (бледность, слабость, одышка) из-за постоянной микрокровопотери.
- Необъяснимая потеря веса.
Неврологические симптомы (мозговые):
Зависят от размера и локализации опухоли (чаще мозжечок при типе 2 или полушария при типе 1):
- Упорные головные боли, которые распирают изнутри и плохо снимаются анальгетиками.
- Нарушение координации движений, шаткость походки, неловкость в руках (особенно при медуллобластоме).
- Изменения зрения (двоение в глазах, выпадение полей зрения).
- Изменение личности, снижение памяти, заторможенность.
КРАСНЫЕ ФЛАГИ (необходима срочная помощь):
- Эпилептические припадки (судороги тела с потерей или без потери сознания).
- Утренняя рвота, приносящая облегчение головной боли, часто фонтаном (признак высокого внутричерепного давления).
- Внезапная слабость или паралич в руке/ноге.
- Профузное кровотечение из прямой кишки.
Ключевые выводы:
- 1. Симптомы складываются из признаков поражения кишечника и мозга.
- 2. Полипы кишечника могут долго не давать симптомов, проявляясь только анемией.
- 3. Утренняя рвота в сочетании с головной болью - грозный признак роста опухоли мозга, требующий немедленного МРТ.
РАЗДЕЛ 5 - ЧТО ДЕЛАТЬ: ПОШАГОВЫЙ ПЛАН ПАЦИЕНТА
Если у вас в семье были случаи раннего рака кишечника, САП или опухолей мозга, либо у вас уже выявлены множественные полипы, необходимо действовать по четкому алгоритму.
Когда нельзя ждать (обращение в тот же день):
Любые острые неврологические нарушения (нарушение речи, перекос лица, судороги, нарушение сознания) или признаки внутреннего кровотечения (черный стул, резкая слабость с падением давления). Вызывайте скорую помощь!
Пошаговый план до визита к врачу:
- 1. Соберите семейный анамнез: Опросите родственников (включая бабушек/дедушек и тетей/дядей). Кто, в каком возрасте и от какого вида рака лечился? Запишите это.
- 2. Запишитесь к врачу-генетику и гастроэнтерологу: Это два главных специалиста на начальном этапе.
- 3. Подготовьте документы: Соберите все предыдущие результаты колоноскопий, гистологические заключения (биопсии полипов), диски МРТ (если делали).
- 4. Сдайте базовые анализы (опционально): Общий анализ крови (для проверки на анемию), анализ кала на скрытую кровь (если еще не делали колоноскопию).
- 5. Не принимайте обезболивающие бесконтрольно: Если болит живот или голова, постоянный прием НПВС (ибупрофен, нимесулид) может замаскировать симптомы или спровоцировать желудочно-кишечное кровотечение.
Что допустимо самостоятельно:
- Переход на легкоусвояемую диету без грубой клетчатки и острых специй (если есть боли в кишечнике).
- Обращение в специализированные пациентские сообщества для психологической поддержки.
- Ведение дневника симптомов (частота стула, наличие крови, характер головных болей).
Чего категорически нельзя делать:
- Лечить полипы народными средствами (клизмы с травами, сода, перекись водорода). Это бесполезно при генетической мутации и ведет к потере драгоценного времени, пока полип перерождается в рак.
- Отказываться от профилактической операции, если ее настаивает провести онкоконсилиум. При САП-ассоциированном синдроме Тюрко риск рака к 40 годам достигает 100%.
- Делать МРТ без контраста при подозрении на опухоль мозга (контрастирование обязательно для точной оценки очагов).
Ключевые выводы:
- 1. Сбор точного семейного анамнеза - половина успеха в диагностике.
- 2. Игнорирование симптомов и самолечение при генетических полипозах фатальны.
- 3. При появлении «красных флагов» необходимо экстренное обращение в стационар.
РАЗДЕЛ 6 - ДИАГНОСТИКА
Диагноз «Синдром Тюрко» ставится на стыке трех дисциплин: гастроэнтерологии, нейрохирургии/неврологии и генетики [4].
Как ставится диагноз: Заподозрить синдром врач может, если у молодого пациента с опухолью мозга в анамнезе (или у родственников) есть множественные полипы кишечника, или наоборот.
Инструментальные методы:
- Видеоколоноскопия высокого разрешения с узкоспектральной визуализацией (NBI): Золотой стандарт. Позволяет увидеть полипы, оценить их количество, размер и сразу взять биопсию (кусочек ткани) для гистологии.
- ЭГДС (гастроскопия): Обязательна, так как полипы при данных мутациях могут расти также в желудке и двенадцатиперстной кишке.
- МРТ головного мозга с внутривенным контрастированием: Необходимо для выявления глиом и медуллобластом, оценки их связи с окружающими тканями и планирования биопсии/операции.

Лабораторные и генетические анализы:
- Секвенирование нового поколения (NGS): Поиск мутаций в генах APC, MLH1, MSH2, MSH6, PMS2. Это главный анализ, подтверждающий природу заболевания и определяющий тип синдрома Тюрко.
- Иммуногистохимия (ИГХ) ткани полипа или опухоли: Позволяет определить дефицит белков системы репарации (признак синдрома Линча).
- Общеклинические анализы крови (выявление анемии).
Дифференциальная диагностика (с чем путают):
- Спорадический (случайный) колоректальный рак + случайная опухоль мозга (без генетической связи).
- Синдром Пейтца-Егерса или ювенильный полипоз (другие типы полипов - гамартомные, а не аденоматозные).
- Синдром Гарднера (как упоминалось выше).
На что смотреть при выборе клиники: Лечение синдрома Тюрко должно проходить только в крупных федеральных или региональных многопрофильных онкологических центрах. Клиника должна обладать сильной молекулярно-генетической лабораторией, отделением нейроонкологии и колопроктологии.
Ключевые выводы:
- 1. Диагноз невозможен без генетического тестирования (NGS-панель).
- 2. Золотой стандарт обследования - тотальная колоноскопия + МРТ головного мозга с контрастом.
- 3. Диагностикой и ведением пациента должна заниматься мультидисциплинарная команда в крупном центре.
РАЗДЕЛ 7 - МЕТОДЫ ЛЕЧЕНИЯ
Поскольку устранить генетическую мутацию во всех клетках организма современной медицине пока не под силу, лечение синдрома Тюрко направлено на удаление уже возникших опухолей и предотвращение развития рака [5].
Таблица подходов к лечению:
| Орган-мишень | Консервативное/Эндоскопическое лечение | Хирургическое лечение (основное) | Системное лечение (Онкология) |
|---|---|---|---|
| Толстая кишка | Эндоскопическая полипэктомия (при типе 1, если полипов мало). | Тотальная колпроктэктомия (полное удаление кишки) с формированием резервуара из тонкой кишки. Показана при ковровом полипозе (тип 2). | Химиотерапия или таргетная/иммунотерапия (если уже развился колоректальный рак с метастазами). |
| Головной мозг | Медикаментозное снятие отека мозга (кортикостероиды), противосудорожные препараты (симптоматически). | Нейрохирургическая операция: максимально безопасное удаление (резекция) опухоли мозга. | Лучевая терапия и системная химиотерапия (обязательный этап после удаления глиобластомы или медуллобластомы). |
Показания к госпитализации:
- Плановая госпитализация для проведения оперативных вмешательств (удаление кишки, удаление опухоли мозга).
- Экстренная госпитализация при осложнениях: острая кишечная непроходимость (опухоль перекрыла просвет), желудочно-кишечное кровотечение, резкое нарастание неврологического дефицита (отек мозга).
Критерии успешного лечения:
- Отсутствие рецидива опухоли мозга по данным контрольных МРТ.
- Устранение риска рака кишечника (после радикального удаления кишки) или контроль роста полипов (при эндоскопическом удалении).
- Сохранение приемлемого качества жизни.
Важно: При типе 2 (мутация APC) профилактическое удаление всей толстой кишки в молодом возрасте - это не "крайняя мера", а единственный способ спасти жизнь пациента от неизбежного рака. Решение принимается консилиумом врачей.
Ключевые выводы:
- 1. Основа лечения - хирургическое вмешательство (нейрохирургия и колопроктология).
- 2. При множественном полипозе профилактическое удаление кишки спасает жизнь.
- 3. Опухоли мозга требуют комплексного подхода: операция + лучевая + химиотерапия.
РАЗДЕЛ 8 - ОСОБЫЕ ГРУППЫ ПАЦИЕНТОВ
Дети и подростки
Синдром Тюрко 2 типа (на фоне мутации APC) бьет преимущественно по детям. Медуллобластома чаще всего диагностируется в возрасте от 5 до 15 лет. Особенности:
- Симптомы могут маскироваться под школьную усталость, проблемы со зрением, укачивание в транспорте (рвота).
- При выявлении медуллобластомы у ребенка обязательно назначается генетический тест и колоноскопия для проверки на полипоз (даже если живот не болит) [6].

Беременные и планирование семьи
Если у человека диагностирован синдром Тюрко, перед планированием беременности необходима консультация репродуктолога-генетика. Современная медицина предлагает преимплантационное генетическое тестирование (ПГТ-М) в рамках ЭКО: эмбрион проверяется на наличие дефектного гена до переноса в матку, что гарантирует рождение здорового ребенка без этого синдрома.
Иммунодефицит и сопутствующие заболевания
Наличие тяжелого сахарного диабета или иммунодефицита усложняет проведение массивных хирургических вмешательств и химиотерапии, повышая риски инфекционных осложнений. Требуется тщательная предоперационная подготовка.
Ключевые выводы:
- 1. Медуллобластома у ребенка - повод проверить кишечник на полипы (и наоборот).
- 2. Процедура ЭКО с генетическим тестированием эмбриона позволяет прервать передачу болезни в семье.
- 3. Лечение детей должно проходить строго в центрах детской нейроонкологии.
РАЗДЕЛ 9 - ЧАСТЫЕ ОШИБКИ ПАЦИЕНТОВ
При редких генетических синдромах цена ошибки очень высока. Вот наиболее частые и опасные заблуждения:
1. «У меня ничего не болит, значит, рака и полипов нет».
- Что делают: Отказываются от плановой колоноскопии.
- Механизм вреда: Полипы толстой кишки не имеют нервных окончаний. Они не болят, даже когда перерождаются в рак. Симптомы появляются только на 3-4 стадии, когда опухоль прорастает в соседние органы или перекрывает просвет кишки.
2. Лечение постоянных головных болей у остеопатов и массажистов.
- Что делают: Месяцами ходят на массаж шейно-воротниковой зоны при утренних головных болях с тошнотой.
- Механизм вреда: Упускается время активного роста опухоли мозга. Массаж и физиотерапия могут спровоцировать усиление внутричерепного давления.
3. Отказ от профилактической колэктомии из-за страха стомы.
- Что делают: Пациенты с ковровым полипозом (сотни полипов) отказываются от операции, надеясь, что полипы "рассосутся" или их можно удалить по одному.
- Механизм вреда: При мутации APC риск рака стремится к 100%. Современная хирургия часто позволяет сформировать резервуар из тонкой кишки, избегая постоянной стомы (мешка на животе), но промедление приводит к инкурабельному (неизлечимому) раку.
4. Сокрытие семейного анамнеза от врачей.
- Что делают: Не рассказывают гастроэнтерологу, что у брата или отца была опухоль мозга.
- Механизм вреда: Врач воспринимает полипы как стандартную возрастную проблему и назначает контроль через 3-5 лет, в то время как при синдроме Тюрко контроль нужен ежегодно.
5. Поиск альтернативного лечения опухолей мозга.
- Что делают: Используют диеты (например, голодание или строгую кето-диету как "лечение"), грибы, настойки вместо операции и химии.
- Механизм вреда: Агрессивные опухоли (глиобластома, медуллобластома) удваивают свой объем за недели. Альтернативная медицина приводит к быстрому летальному исходу от отека мозга.
Ключевые выводы:
- 1. Отсутствие симптомов не означает отсутствие болезни.
- 2. Игнорирование генетической природы болезни и отказ от превентивной хирургии ведет к развитию рака.
- 3. Утаивание семейной истории болезней лишает врачей возможности поставить правильный диагноз.
РАЗДЕЛ 10 - ПРОФИЛАКТИКА
Говорить о классической профилактике (как при инфекциях) в случае генетического заболевания нельзя. Однако медицина предлагает надежные способы контроля.
Первичная профилактика (предотвращение болезни у потомства):
Единственный метод - упомянутое выше ПГТ-М (преимплантационное генетическое тестирование) при процедуре ЭКО. Это позволяет парам с подтвержденной мутацией родить ребенка без гена синдрома Тюрко.
Вторичная профилактика (предотвращение рака и тяжелых осложнений):
Основана на жесточайшем пожизненном диспансерном скрининге носителей мутации (даже до появления симптомов) [7].
Примерный график скрининга (может корректироваться врачом):
- С 10-12 лет (или на 5 лет раньше самого раннего рака в семье): Ежегодная колоноскопия (при мутации APC) или раз в 1-2 года (при мутации синдрома Линча).
- С 10-15 лет: ЭГДС (гастроскопия) каждые 1-3 года для контроля желудка и ДПК.
- С детского возраста (по назначению генетика/невролога): МРТ головного мозга с контрастом каждые 1-2 года.
- УЗИ щитовидной железы ежегодно (при синдроме Линча/САП часто бывают сопутствующие патологии).

Бытовые меры: Пациентам с удаленной толстой кишкой требуется специальная диета, богатая белком, с контролем водно-электролитного баланса (так как толстая кишка отвечает за всасывание воды).
Ключевые выводы:
- 1. Первичная профилактика возможна только на этапе планирования беременности (ЭКО + ПГТ-М).
- 2. Вторичная профилактика - это ежегодные эндоскопия и МРТ по расписанию.
- 3. Диспансерное наблюдение ведется пожизненно.
FAQ (Частые вопросы)
1. Синдром Тюрко заразен?
Нет. Это полностью генетическое заболевание, обусловленное поломкой в ДНК. Заразиться им бытовым, воздушно-капельным или половым путем невозможно [8].
2. Можно ли жить полноценной жизнью с этим диагнозом?
Да, при условии раннего выявления. Если полипы удаляются превентивно (или проводится операция на кишке до развития рака), а опухоль мозга обнаружена на ранней стадии и успешно пролечена, пациенты живут десятилетиями, работают и создают семьи.
3. В чем разница между синдромом Тюрко и синдромом Гарднера?
Оба синдрома вызывают полипоз кишечника. Но при синдроме Тюрко полипоз сочетается с опухолями мозга, а при синдроме Гарднера - с доброкачественными опухолями костей (остеомами) и мягких тканей (фибромами, десмоидами) [1].
4. Обязательно ли удалять всю толстую кишку?
Если подтвержден тип 2 (мутация гена APC), то да, это практически неизбежно. Кишка покрывается ковром из тысяч полипов, удалить их по одному невозможно, и риск рака приближается к 100%. Хирурги стараются провести операцию так, чтобы сохранить пациенту возможность естественного стула без постоянной стомы.
5. Какие именно гены нужно проверить?
В первую очередь проверяются гены системы мисматч-репарации (MLH1, MSH2, MSH6, PMS2) и ген APC. Анализ обычно назначает врач-генетик в виде комплексной панели на наследственные опухолевые синдромы.
6. У меня нет симптомов, но у отца был синдром Тюрко. Нужно ли мне проверяться?
Обязательно. Риск того, что вы унаследовали мутацию, составляет 50%. Вам необходимо срочно обратиться к генетику для тестирования. До появления первых симптомов опухоли могут развиваться годами [9].
7. Передается ли синдром через поколение (от деда к внуку, минуя отца)?
Как правило, наследование идет аутосомно-доминантно - то есть непрерывно из поколения в поколение. Однако, изредка ген может проявляться очень слабо в одном поколении (неполная пенетрантность), создавая иллюзию «пропуска». Поэтому проверяться должны все кровные родственники первой линии.
Источники и литература
- Источник: Turcot Syndrome - StatPearls - NCBI Bookshelf - URL (дата обращения: 18.02.2026)
- Источник: Turcot Syndrome - Cancer.Net (ASCO) - URL (дата обращения: 18.02.2026)
- Источник: OMIM (Online Mendelian Inheritance in Man) - Turcot Syndrome - URL (дата обращения: 18.02.2026)
- Источник: Клинические рекомендации Минздрава РФ «Семейный аденоматозный полипоз» - URL (дата обращения: 18.02.2026)
- Источник: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Colorectal - URL (дата обращения: 18.02.2026)
- Источник: Orphanet: Turcot syndrome - URL (дата обращения: 18.02.2026)
- Источник: American Gastroenterological Association (AGA) Guidelines on FAP and Turcot - URL (дата обращения: 18.02.2026)
- Источник: WHO Fact sheet: Cancer (Genetic predispositions) - URL (дата обращения: 18.02.2026)
- Источник: Turcot Syndrome Overview - Medscape - URL (дата обращения: 18.02.2026)
Как подготовлена статья:
Автор: медицинский редактор портала med-oko.ru, врач со стажем 15 лет.
Статья написана в соответствии с редакционной политикой.
Материал пересматривается при обновлении источников, изменении клинических подходов или не реже одного раза в год.
Дисклеймер: Материал предназначен для информирования и не является диагнозом, назначением лечения или заменой очного осмотра. При упорных головных болях, нарушении координации, примеси крови в кале или изменении характера стула обратитесь к врачу.