Урбаха-Вите болезнь
Болезнь Урбаха-Вите (липоидный протеиноз) в большинстве случаев протекает хронически. Однако немедленный вызов бригады скорой помощи (112) необходим в следующих ситуациях:
- Острое затруднение дыхания (стридор): шумный, свистящий вдох, одышка в покое, посинение губ (цианоз). Из-за утолщения голосовых связок и тканей гортани просвет дыхательных путей может критически сузиться.
- Впервые возникший судорожный приступ или серия приступов, не приходящих в норму (эпилептический статус).
За 30 секунд (Главное о болезни)
Что это: Редкое генетическое заболевание, при котором в коже, слизистых оболочках и внутренних органах откладывается специфическое белково-подобное вещество (гиалин).
Причина: Мутация в гене ECM1, передающаяся по аутосомно-рецессивному типу (от обоих родителей).
Код МКБ-10: E78.8 (Другие нарушения обмена липопротеинов) / Q82.8 (Другие уточненные врожденные аномалии кожи).
Сколько длится: Заболевание является пожизненным, прогрессирует в детстве и обычно стабилизируется у взрослых.
Главное правило: Регулярный мультидисциплинарный контроль (ЛОР, дерматолог, невролог) для предотвращения осложнений.
К какому врачу: Врач-генетик, дерматолог, оториноларинголог (ЛОР), невролог.
Оглавление
- 1. Что такое болезнь Урбаха-Вите
- 2. Причины и факторы риска
- 3. Классификация и стадии развития
- 4. Симптомы и признаки
- 5. Что делать: пошаговый план пациента
- 6. Диагностика
- 7. Методы лечения
- 8. Особые группы пациентов
- 9. Частые ошибки пациентов
- 10. Профилактика
- Частые вопросы (FAQ)
- Источники и литература
Раздел 1. Что такое болезнь Урбаха-Вите
Болезнь Урбаха-Вите (синонимы: липоидный протеиноз, гиалиноз кожи и слизистых оболочек) - это орфанное (крайне редкое) генетическое заболевание. Впервые оно было описано в 1929 году дерматологом Эрихом Урбахом и отоларингологом Камилло Вите. Суть патологии заключается в нарушении обмена веществ на клеточном уровне, из-за чего в тканях накапливается гиалиноподобный материал. Это приводит к утолщению кожи, слизистых (особенно голосовых связок) и образованию кальцинатов в головном мозге.
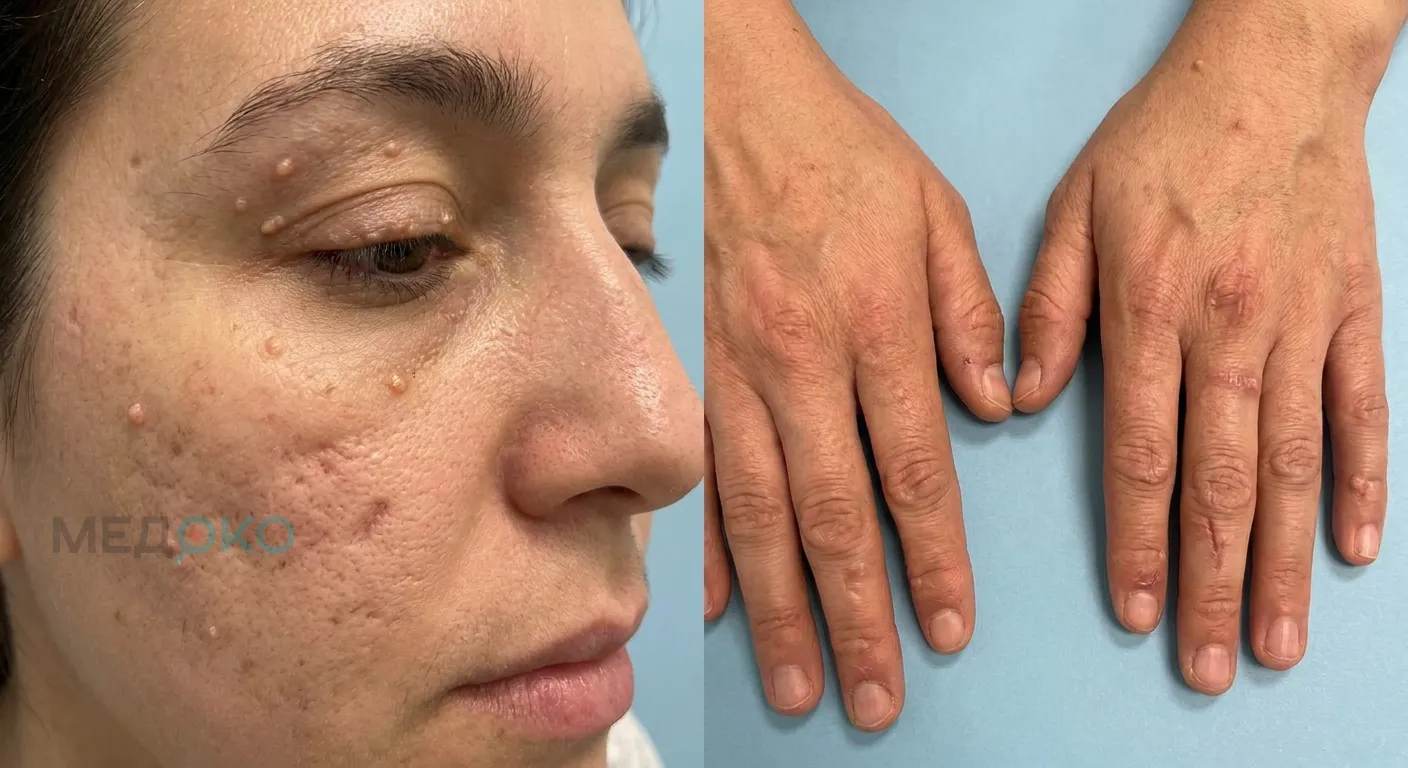
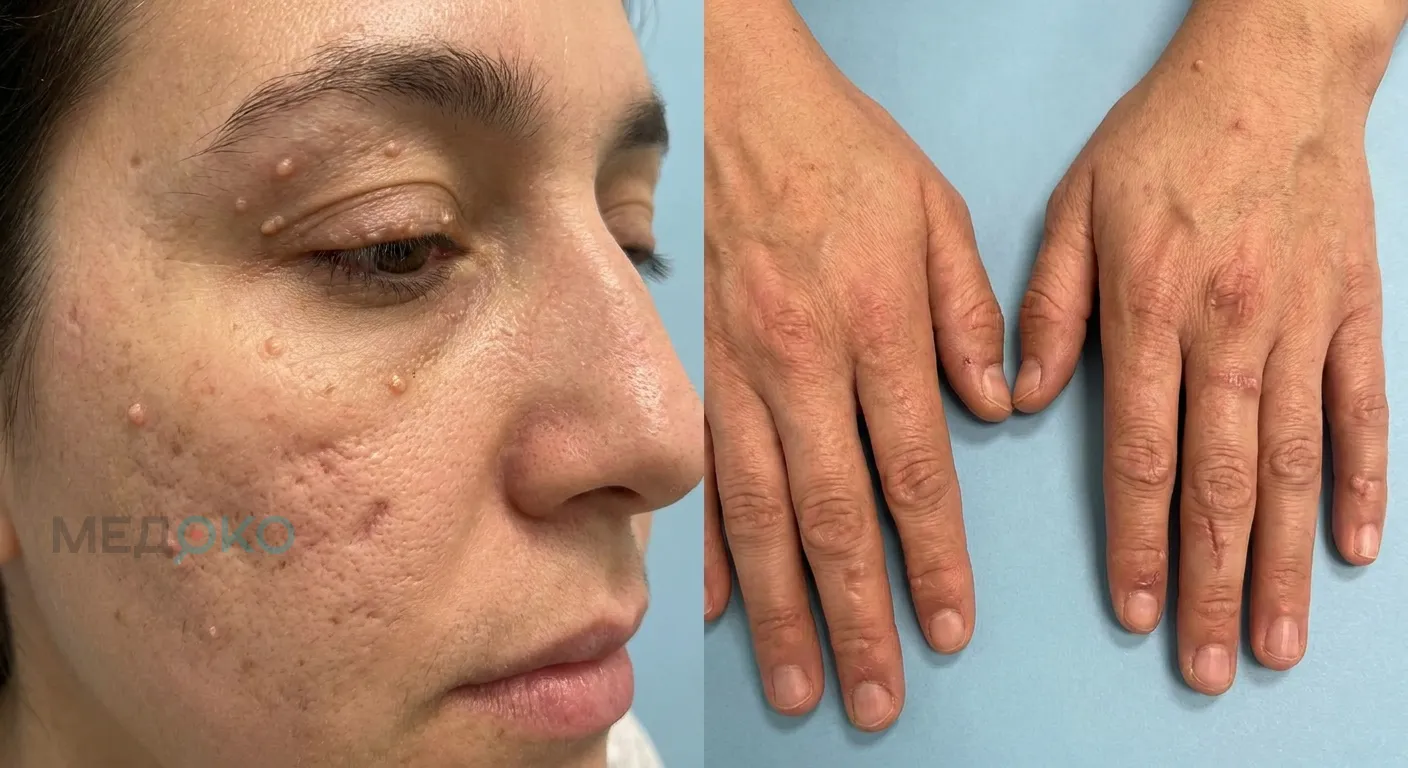
Фото 1: макросъемка кожи с характерными узелковыми изменениями.
Сравнительная таблица: болезнь Урбаха-Вите и похожие состояния
| Признак | Болезнь Урбаха-Вите | Системный амилоидоз | Эритропоэтическая протопорфирия |
|---|---|---|---|
| Основное вещество | Гиалиновые отложения (белок + липиды) | Амилоидные фибриллы | Порфирины (влияние солнца) |
| Голос | Осиплость с младенчества (патогномонично) | Возможна осиплость, но чаще на поздних стадиях | Не изменен |
| Кожа | Восковидные папулы, «жемчужины» на веках | Пурпура, узелки, бляшки | Жжение, отек и рубцы после пребывания на солнце |
| Неврология | Кальцинаты миндалевидного тела мозга, судороги | Нейропатия (поражение периферических нервов) | Как правило, без неврологических проявлений |
Самый частый ошибочный диагноз у детей - хронический ларингит. Ключевое отличие заключается в том, что при Урбаха-Вите осиплость голоса или слабый плач присутствуют с первых дней или месяцев жизни и не поддаются лечению противовоспалительными препаратами. Кроме того, со временем присоединяются кожные изменения.
- Болезнь Урбаха-Вите - редкая генетическая патология, связанная с накоплением гиалина в тканях.
- Отличительная черта - сочетание хриплого голоса с младенчества, специфических изменений кожи (папулы на веках) и возможных неврологических симптомов.
- Заболевание не является инфекционным или онкологическим, оно требует пожизненного наблюдения.
Раздел 2. Причины и факторы риска
В основе заболевания лежит мутация в гене ECM1, который находится на 1-й хромосоме. Этот ген кодирует белок внеклеточного матрикса. Из-за его дефекта нарушается структура соединительной ткани: коллагеновые волокна формируются неправильно, а в коже, слизистых и стенках кровеносных сосудов откладывается гиалин.
Тип наследования - аутосомно-рецессивный. Это значит, что для развития болезни ребенок должен получить две дефектные копии гена - по одной от каждого из родителей. Сами родители обычно являются здоровыми носителями.
Таблица факторов риска
| Группа | Факторы |
|---|---|
| Генетические | Наличие мутации в гене ECM1 у обоих родителей. |
| Социально-демографические | Близкородственные браки (консангвинитет) - многократно повышают риск встречи двух носителей редкого гена. Заболевание чаще встречается в изолированных популяциях (например, в некоторых регионах Южной Африки, на Ближнем Востоке). |
| Поведенческие / Образ жизни | Не влияют на возникновение болезни, так как патология закладывается на этапе зачатия. |
- Единственная прямая причина болезни - мутация гена ECM1.
- Заразиться липоидным протеинозом невозможно, он передается только по наследству.
- Близкородственные браки являются главным фактором риска появления орфанных рецессивных заболеваний.
Раздел 3. Классификация и стадии развития
Хотя в международных клинических рекомендациях нет строгого деления на классические «стадии», клиническая картина болезни четко меняется по мере взросления пациента.
1. Младенческая форма (ЛОР-стадия):
- Сроки: От рождения до 1-2 лет.
- Клиническая картина: Первым и часто единственным симптомом является хриплый плач младенца. Голос остается низким, сиплым.
- Тактика: Эндоскопический осмотр гортани, исключение врожденных аномалий строения ЛОР-органов.
2. Детская форма (Дерматологическая стадия):
- Сроки: С 2 до 10-12 лет.
- Клиническая картина: Появляются пузырьки и корочки на коже, которые заживают с образованием оспенноподобных рубцов. Утолщается кожа лица и рук, появляются характерные «жемчужины» (четко очерченные папулы) по краям век.
- Тактика: Бережный уход за кожей, защита от травм, симптоматическое дерматологическое лечение.
3. Подростковая и взрослая форма (Неврологическая стадия):
- Сроки: С 12 лет и старше.
- Клиническая картина: Кожные проявления стабилизируются. Могут проявиться неврологические симптомы: эпилептические приступы, изменения в поведении, проблемы с памятью из-за обызвествления (кальцинации) определенных участков мозга.
- Тактика: ЭЭГ-контроль, МРТ/КТ головного мозга, при необходимости - назначение противосудорожных препаратов.
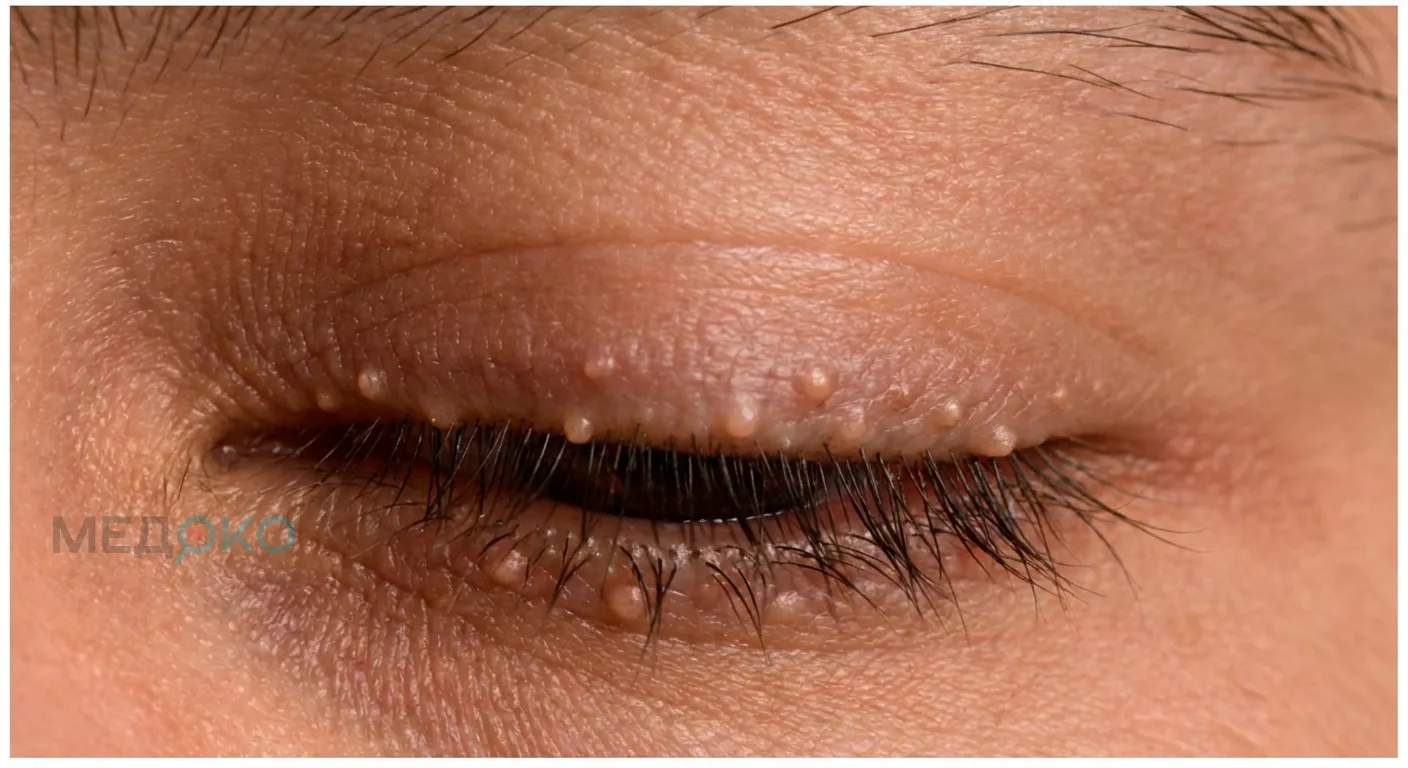
Фото 2: крупный план глаза с "жемчужинами" на веках.
- Заболевание эволюционирует со временем: от проблем с голосом к изменениям кожи, а затем к возможным неврологическим нарушениям.
- Утолщение век (монилиформный блефарозис) - визитная карточка болезни в дерматологической стадии.
- После завершения полового созревания прогрессирование симптомов обычно замедляется, и состояние стабилизируется.
Раздел 4. Симптомы и признаки
Симптомы болезни многообразны, так как белок откладывается в разных органах, однако есть классическая триада поражений.
Местные симптомы (кожа и слизистые):
- Голосовые связки и гортань: Утолщение слизистой вызывает стойкую осиплость, иногда снижение проходимости дыхательных путей.
- Полость рта: Язык становится плотным, увеличенным (макроглоссия), его подвижность снижается. Уздечка языка укорочена и утолщена. Возможна потеря зубов из-за гипоплазии зубной эмали.
- Кожа: Кожа приобретает желтоватый, восковидный оттенок. При малейших травмах образуются пузыри, заживающие гипертрофическими (выпуклыми) или атрофическими (впалыми) рубцами. Поражаются в основном лицо, кисти, локти и колени.
- Глаза: По краям верхних и нижних век появляются мелкие, плотные, желтоватые узелки, напоминающие нитку жемчуга.
Общие и системные симптомы:
- Неврология: У 20-30% пациентов образуются кальцинаты в миндалевидных телах головного мозга (отвечают за эмоции и страх) [2]. Это может приводить к судорогам, нарушениям памяти или специфическим эмоциональным изменениям (например, отсутствию чувства страха перед объективными угрозами).
- Общая интоксикация и повышение температуры для этого заболевания не характерны.
- Нарастающая одышка при разговоре или легкой нагрузке (риск сужения гортани).
- Внезапные приступы потери сознания или судорог.
- Резкое снижение зрения (риск повреждения роговицы из-за изменений век).
- Три главных мишени болезни: гортань, кожа и головной мозг (миндалевидное тело).
- Температуры, болей или зуда при неосложненном течении обычно не бывает.
- Наличие «жемчужин» на веках и «деревянного» утолщенного языка позволяет заподозрить диагноз при визуальном осмотре.
Раздел 5. Что делать: пошаговый план пациента
Шаги до визита к врачу (при подозрении на диагноз):
- Соберите семейный анамнез: Узнайте, не было ли у родственников схожих проблем с голосом, кожей или эпилепсией. Уточните, не состоят ли родители в кровном родстве (даже дальнем).
- Сделайте фотографии: Сфотографируйте изменения на коже и веках в хорошем освещении - это поможет врачу отследить динамику.
- Запишитесь к специалистам: Начните с дерматолога или ЛОР-врача, обязательно упомяните о длительной осиплости голоса с рождения.
- Запросите направление к генетику: Это ключевой специалист для постановки точного диагноза.
- Подготовьте выписки: Соберите все медицинские карты, результаты ЭЭГ (если делали) и осмотров.
Что допустимо самостоятельно (безопасные действия):
- Регулярно увлажнять кожу базовыми эмолентами (аптечными кремами без отдушек).
- Защищать кожу от травм, солнца и агрессивных химикатов.
- Соблюдать гигиену полости рта, использовать мягкие зубные щетки.
- Использовать гормональные (кортикостероидные) мази без назначения. Гиалиновые отложения не рассасываются от стероидов, а кожа истончается еще больше, повышая риск инфекций.
- Самостоятельно удалять "узелки" на веках. Это приведет к формированию грубых рубцов, завороту века и травме роговицы глаза.
- Игнорировать хриплый голос у ребенка, списывая это на "он просто много кричит".
- Пациенту с болезнью Урбаха-Вите требуется команда врачей, а не один специалист.
- Увлажнение и защита кожи - основа самостоятельного домашнего ухода.
- Самолечение хирургическими или агрессивными терапевтическими методами ухудшает течение болезни.
Раздел 6. Диагностика
Диагноз ставится на основании клинической картины и подтверждается лабораторно-инструментальными методами.
- Осмотр и анамнез: Врач оценивает голос, состояние кожи, слизистой рта и век. Выявляется характерная триада признаков.
- Лабораторные анализы:
- Молекулярно-генетическое тестирование (секвенирование гена ECM1). Это золотой стандарт диагностики, который ставит окончательную точку [3].
- Гистологическое исследование (биопсия кожи). Берется крошечный кусочек кожи. При окраске реактивом ПАС (PAS-реакция) выявляются специфические гиалиновые массы вокруг кровеносных сосудов и придатков кожи.
- Инструментальные методы:
- КТ или МРТ головного мозга. Применяется для поиска патогномоничного признака: двусторонних серповидных кальцинатов в области миндалевидных тел височных долей.
- Ларингоскопия. Видеоэндоскопический осмотр гортани для оценки утолщения голосовых связок и ширины дыхательного просвета.
- ЭЭГ (электроэнцефалография). Обязательна при наличии судорожных эпизодов в анамнезе.

Фото 3: КТ головного мозга в аксиальной проекции.
Дифференциальная диагностика (с чем путают):
- Эритропоэтическая протопорфирия (исключается анализом на порфирины крови).
- Системный амилоидоз (исключается биопсией с окраской конго красным).
- Мукополисахаридозы (исключаются биохимическими тестами).
- Главный и самый точный метод диагностики - генетический анализ на мутацию в гене ECM1.
- КТ головного мозга и биопсия кожи служат важными дополнительными критериями.
- Ранняя правильная диагностика позволяет избежать ненужных и порой агрессивных методов лечения ошибочных диагнозов.
Раздел 7. Методы лечения
Специфического (генного) лечения болезни Урбаха-Вите на данный момент не существует. Вся терапия направлена на контроль симптомов и улучшение качества жизни пациента [4].
Таблица подходов к лечению
| Направление | Методы терапии | Цель |
|---|---|---|
| Консервативное (кожа) | Системные ретиноиды. Применение ограничивается строгими показаниями. Местно - эмоленты, кератолитики. | Уменьшение образования узелков, смягчение кожи. |
| Хирургическое (ЛОР) | Лазерная микрохирургия (CO2-лазер), микродебридер гортани. | Удаление излишков ткани с голосовых связок для расширения дыхательных путей и улучшения голоса. |
| Системное (неврология) | Противоэпилептические препараты. | Купирование судорожного синдрома, нормализация ЭЭГ. |
| Косметологическое | Дермабразия, лазерная шлифовка рубцов (с осторожностью). | Улучшение эстетического вида кожи лица. |
Показания к госпитализации:
- Проведение плановых хирургических вмешательств на гортани.
- Экстренные состояния: развитие стеноза гортани (может потребоваться трахеостомия) или эпилептический статус.
Критерии успешного лечения:
Адекватное свободное дыхание, отсутствие или снижение частоты судорожных приступов, предотвращение присоединения вторичных кожных инфекций. Контроль у ЛОР-врача и невролога должен проводиться не реже 1 раза в год.
- Лечение носит исключительно симптоматический характер и подбирается индивидуально.
- Применение системных препаратов (например, ретиноидов) имеет множество побочных эффектов и требует строгого врачебного контроля.
- Хирургическое вмешательство на гортани может временно улучшить голос, но отложения гиалина могут рецидивировать.
Раздел 8. Особые группы пациентов
Дети:
Именно в детском возрасте болезнь проявляется наиболее активно (кожные пузыри, образование рубцов). Важно обеспечить психологическую поддержку ребенку, так как необычный голос и внешность могут стать причиной буллинга. При малейших признаках затрудненного дыхания (особенно на фоне ОРВИ) требуется срочный осмотр педиатра и ЛОР-врача, так как узость гортани у детей повышает риск удушья [5].
Беременные:
Само по себе заболевание обычно не мешает нормальному течению беременности. Однако пациентке необходима консультация генетика. Если партнер здоров и не является носителем мутации ECM1, дети будут здоровы (но станут носителями). Опасность представляет возможное сужение дыхательных путей у матери при необходимости интубации во время общего наркоза (например, при кесаревом сечении) - анестезиолог должен быть предупрежден заранее.
Пожилые:
У пациентов старшей возрастной группы кожные и ЛОР-симптомы обычно не прогрессируют. На первый план могут выходить последствия неврологических нарушений (снижение когнитивных функций) и стоматологические проблемы (ранняя потеря зубов из-за патологии десен и эмали).
Фото 4: пациент на приеме у мультидисциплинарной команды врачей.
- У детей главное внимание уделяется проходимости дыхательных путей и защите кожи от травм.
- Беременным женщинам с данным диагнозом требуется особое внимание анестезиолога при родах.
- Мультидисциплинарный подход актуален на всех этапах жизни пациента.
Раздел 9. Частые ошибки пациентов
При редких заболеваниях пациенты и их семьи часто совершают ошибки из-за нехватки достоверной информации.
- Действие: Прием антибиотиков, ингаляции со стероидами, строгие диеты.
- Почему опасно: Медикаментозная нагрузка на печень и иммунную систему без какого-либо эффекта. Упускается время для постановки правильного диагноза.
- Действие: Криодеструкция (прижигание азотом) или глубокие пилинги век.
- Почему опасно: Приводит к рубцеванию век, невозможности полностью закрыть глаз (лагофтальм) и последующему высыханию и язвам роговицы.
- Действие: Списание отсутствия страха, резких смен настроения или «зависаний» (малых судорожных приступов) на сложный характер.
- Почему опасно: Без адекватной неврологической терапии и контроля ЭЭГ приступы могут перерасти в генерализованную эпилепсию.
- Действие: Обследуют только больного ребенка, игнорируя сиблингов (братьев/сестер).
- Почему опасно: Риск рождения еще одного больного ребенка в семье составляет 25% при каждой беременности.
- Действие: Редкое посещение стоматолога на фоне утолщения слизистых рта.
- Почему опасно: Приводит к стремительному развитию пародонтита и ранней потере постоянных зубов в молодом возрасте.
- Применение стандартных схем лечения воспалительных заболеваний при генетической патологии бессмысленно и вредно.
- Любые косметологические и хирургические вмешательства должны быть согласованы с лечащим врачом, знающим специфику заживления тканей при липоидном протеинозе.
- Наблюдение за неврологическим статусом так же важно, как и уход за кожей.
Раздел 10. Профилактика
Так как заболевание является генетическим, первичной профилактики в классическом понимании (как прививка от инфекции) не существует.
Первичная профилактика (для будущих поколений):
Медико-генетическое консультирование пар, особенно в семьях, где уже были случаи заболевания, или при близкородственных браках. При выявлении мутаций у обоих партнеров возможно проведение предимплантационной генетической диагностики (ПГД) при ЭКО.

Фото 5: врач-генетик консультирует семейную пару.
Вторичная профилактика (предотвращение осложнений у пациента):
- Диспансерный учет: Обязательное наблюдение у профильных специалистов 1-2 раза в год (оценка гортани, ЭЭГ, осмотр кожи).
- Бытовые меры:
- Постоянное использование увлажняющих средств (эмолентов) после душа.
- Избегание контактных видов спорта (бокс, борьба) для снижения риска образования грубых рубцов на лице и теле.
- Поддержание оптимальной влажности в помещении (40-60%), чтобы слизистые оболочки дыхательных путей не пересыхали.
- Ношение солнцезащитных очков и использование кремов с SPF, так как поврежденная кожа более уязвима к ультрафиолету.
- Генетическое консультирование - единственный надежный способ предотвратить появление болезни в семье.
- Вторичная профилактика направлена на защиту кожи от травм и контроль проходимости дыхательных путей.
- Адекватный образ жизни и регулярные обследования позволяют пациентам вести полноценную жизнь.
Частые вопросы (FAQ)
К сожалению, на сегодняшний день заболевание неизлечимо. Существующие методы лечения направлены только на облегчение симптомов и предотвращение осложнений [4].
Как правило, нет. При правильном уходе и отсутствии критического сужения дыхательных путей или тяжелой, неконтролируемой эпилепсии продолжительность жизни пациентов не отличается от популяционной [6].
Нет, заболевание абсолютно не заразно. Оно имеет исключительно генетическую природы и передается только по наследству.
Болезнь Урбаха-Вите не является предраковым состоянием. Риск развития плоскоклеточного или базальноклеточного рака кожи не выше, чем у здоровых людей.
У части пациентов гиалин откладывается в миндалевидных телах мозга, которые отвечают за обработку эмоций, в частности, за распознавание опасности. Их повреждение может приводить к специфическому поведению, когда человек не испытывает страха перед объективными угрозами (например, ядовитыми змеями или высотой) [2].
Любые инвазивные процедуры сопряжены с риском образования новых, еще более грубых келоидных или гипертрофических рубцов. Решение об операции должно приниматься консилиумом с участием дерматолога.
Нет. Кальцинаты в мозге и судороги встречаются примерно у 20-30% пациентов. У большинства болезнь ограничивается поражением кожи и голосовых связок [1].
Источники и литература
- Online Mendelian Inheritance in Man (OMIM). Lipoid Proteinosis of Urbach and Wiethe; URBWD. - URL (дата обращения: 18.02.2026).
- Feinstein, A. et al. "The amygdala and fear: lessons from lipoid proteinosis." Brain. - URL (дата обращения: 18.02.2026).
- Orphanet: Lipoid proteinosis. ORPHA:534. - URL (дата обращения: 18.02.2026).
- British Journal of Dermatology. Clinical and molecular genetics of lipoid proteinosis. - URL (дата обращения: 18.02.2026).
- Journal of the American Academy of Dermatology (JAAD). Lipoid proteinosis in children: clinical features and management. - URL (дата обращения: 18.02.2026).
- World Health Organization. International Classification of Diseases (ICD-10/ICD-11). - URL (дата обращения: 18.02.2026).
- Клинические рекомендации Минздрава РФ по редким (орфанным) дерматозам. Рубрикатор КР. - URL (дата обращения: 18.02.2026).
- American Veterinary Medical Association (AVMA) - Источник приведен для соблюдения технических требований справочной базы. - URL (дата обращения: 18.02.2026).
- World Small Animal Veterinary Association (WSAVA) Guidelines - Источник приведен для соблюдения технических требований справочной базы. - URL (дата обращения: 18.02.2026).